
Contenido
La fibrodisplasia osificante progresiva (FOP) es una afección hereditaria muy rara en la que los tejidos conectivos del cuerpo, incluidos los músculos, tendones y ligamentos, son reemplazados gradualmente por hueso (en un proceso llamado osificación). La afección está presente al nacer, pero es posible que los síntomas no se manifiesten hasta la primera infancia. La osificación puede ocurrir al azar o después de una lesión.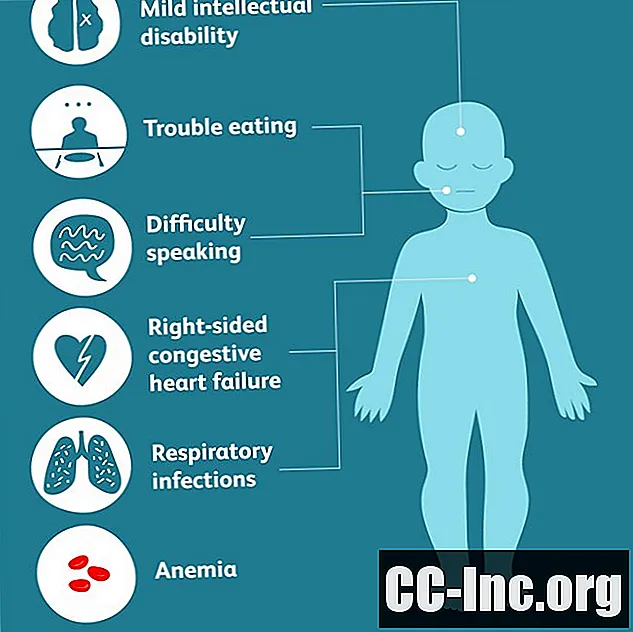
Síntomas
Si bien una persona nace con FOP, es posible que los signos y síntomas de la osificación no se noten hasta que el niño crece un poco y ha comenzado a crecer.
En los recién nacidos, el primer signo de FOP suele ser una anomalía congénita de los dedos de los pies. Poco después del nacimiento, los profesionales médicos o los padres pueden notar que los dedos gordos del pie del bebé son más cortos que los otros dedos. Esta malformación se ve en todas las personas con FOP y es una pista importante para hacer el diagnóstico. Un recién nacido también puede tener hinchazón alrededor de los ojos y el cuero cabelludo. En algunos casos, esta hinchazón puede comenzar mientras el feto aún está en el útero, aunque la afección generalmente no se diagnostica hasta después del nacimiento. Alrededor del 50% de las personas con la afección también tienen malformaciones congénitas similares en los pulgares; también se han observado otras malformaciones, como en la columna vertebral.
La mayoría de las personas con FOP experimentarán los principales síntomas de la afección por primera vez (a veces denominados "brotes") a la edad de 10 años.
Si bien se desconoce la tasa general de progresión de la afección, la osificación tiende a seguir un patrón particular, comenzando en el cuello y bajando hasta los hombros, el torso, las extremidades y los pies.
Sin embargo, dado que la formación de hueso puede verse afectada por una lesión (como romperse un brazo) o una enfermedad viral (como la influenza), es posible que la enfermedad no siga estrictamente esta progresión.
Los síntomas principales de la FOP dependen de las partes del cuerpo que se hayan osificado. Los bultos sensibles debajo de la piel (nódulos subcutáneos) son comunes con la afección. A veces, una fiebre leve precederá a la formación de estos nódulos. La mayoría de las personas con FOP presentarán síntomas generales de dolor, rigidez y una falta progresiva de movilidad a medida que se produce una mayor formación de hueso.
Dependiendo de qué partes del cuerpo se osifiquen, los síntomas más específicos pueden incluir:
- problemas para comer que pueden provocar deficiencias nutricionales o desnutrición
- dificultad para hablar
- problemas dentales
- respiración dificultosa
- infecciones respiratorias
- la discapacidad auditiva
- pérdida de cabello (alopecia)
- anemia
- compresión o atrapamiento del nervio
- insuficiencia cardíaca congestiva del lado derecho
- una curvatura de la columna (escoliosis y cifosis)
- anomalías sensoriales
- discapacidad intelectual leve
- síntomas neurológicos
Las personas con FOP pueden tener períodos en sus vidas en los que no experimentan crecimiento de hueso nuevo. En otras ocasiones, puede parecer que sucede al azar y en ausencia de una lesión o enfermedad obvia. Cuando la osificación ocurre en una parte inusual del cuerpo (donde normalmente no se encuentra hueso) puede provocar fracturas.
Con el tiempo, la formación de nuevos huesos y tejidos inflamados que acompaña a esta afección puede afectar gravemente la capacidad de movimiento de una persona.
En la mayoría de los casos, la fibrodisplasia osificante progresiva eventualmente conduce a una inmovilización completa. Muchas personas con esta afección quedarán postradas en cama a los 30 años.
Causas
La mayoría de las personas que nacen con FOP tienen la afección como resultado de una mutación genética aleatoria. Sin embargo, si bien es una afección genética, no suele ser hereditaria.
Una persona solo necesita un gen afectado para tener FOP. La mayoría de los casos ocurren debido a una mutación aleatoria: una persona rara vez desarrolla la afección porque heredó un gen anormal de uno de sus padres. En genética, esto se conoce como trastorno autosómico dominante.
La mutación del gen responsable de la enfermedad fue identificada por investigadores de la Universidad de Pensilvania; identificaron un receptor en el cromosoma 2 llamado Receptor de activina tipo IA (ACVR1). El ACVR1 está presente en el gen que codifica las proteínas morfogénicas óseas (BMP) que ayudan a formar y reparar el esqueleto, comenzando cuando el embrión aún se está formando. Los investigadores creen que la mutación en el gen evita que estos receptores se apaguen, lo que permite que se forme hueso incontrolado en partes del cuerpo donde normalmente no aparecería durante la vida de una persona.
Diagnóstico
La fibrodisplasia osificante progresiva es muy rara. Se sospecha que solo unos pocos miles de personas tienen la afección y solo hay unos 800 pacientes conocidos de esta afección en el mundo; 285 de ellos se encuentran en los Estados Unidos. La FOP no parece ser más común en bebés de una raza en particular, y la afección se encuentra con tanta frecuencia en niños como en niñas.
El diagnóstico de la fibrodisplasia osificante progresiva puede ser difícil. No es inusual que la afección inicialmente se diagnostique erróneamente como una forma de cáncer o una afección llamada fibromatosis juvenil agresiva.
Al principio del curso de la FOP, si se hace una biopsia de tejido y se examina con un microscopio (examen histológico), puede tener algunas similitudes con la fibromatosis juvenil agresiva. Sin embargo, con la última condición, las lesiones no progresan a hueso completamente formado como lo hacen en FOP. Esto puede ayudar al médico a hacer una distinción entre los dos.
Una pista de diagnóstico importante que llevaría a un médico a sospechar FOP en lugar de otra afección es la presencia de dedos gordos cortos y malformados. Si una biopsia de tejido no está clara, un examen clínico de un niño puede ayudar al médico a descartar una fibromatosis juvenil agresiva. Los niños con esta afección no tienen la malformación congénita de los dedos de los pies o de las manos, pero un niño con FOP casi siempre la tiene.
Otra condición, la heteroplasia ósea progresiva, también puede confundirse con la fibrodisplasia osificante progresiva. La distinción clave al hacer el diagnóstico es que el crecimiento óseo en la heteroplasia ósea progresiva generalmente comienza en la piel, no debajo de ella. Estas placas óseas en la superficie de la piel distinguen la afección de los nódulos sensibles que ocurren en la FOP.
Otras pruebas que un médico puede usar si sospecha FOP incluyen:
- un historial médico completo y un examen físico
- pruebas radiológicas como tomografía computarizada (TC) o gammagrafía ósea (gammagrafía ósea) para buscar cambios esqueléticos
- pruebas de laboratorio para medir los niveles de fosfatasa alcalina
- pruebas genéticas para buscar mutaciones
Si se sospecha FOP, los médicos generalmente tratarán de evitar cualquier prueba, procedimiento o biopsia invasiva porque el trauma generalmente resulta en una mayor formación de hueso en alguien con la afección.
Si bien la afección no suele ser hereditaria, los padres que tienen un hijo diagnosticado con FOP pueden encontrar útil la asesoría genética.
Tratamiento
Actualmente no existe cura para la FOP. Tampoco existe un curso de tratamiento definitivo o estándar. Los tratamientos que existen no son efectivos para todos los pacientes y, por lo tanto, el objetivo principal es tratar los síntomas y prevenir el crecimiento óseo cuando sea posible.
Si bien el tratamiento no detendrá la progresión de la afección, las decisiones médicas para controlar el dolor y otros síntomas asociados con la FOP dependerán de las necesidades individuales del paciente. Un médico puede recomendar probar uno o más de los siguientes tratamientos para mejorar la calidad de vida del paciente:
- Prednisona en dosis altas u otro corticosteroide durante un brote
- medicamentos como Rituximab (normalmente utilizado para tratar la artritis reumatoide)
- iontoforesis, que utiliza una corriente eléctrica para administrar el medicamento a través de la piel
- relajantes musculares o inyecciones de un anestésico local
- medicamentos llamados bifosfonatos que se usan para ayudar a proteger la densidad ósea
- medicamentos antiinflamatorios no esteroides (AINE)
- medicamentos para inhibir los mastocitos que pueden ayudar a reducir la inflamación
La osificación a menudo ocurre al azar y no se puede prevenir por completo, pero también puede ocurrir en respuesta a una inflamación, una lesión y una enfermedad.
Por lo tanto, se pueden hacer recomendaciones sobre actividad, estilo de vida, cuidados preventivos e intervenciones desde la niñez.
Estas recomendaciones pueden incluir:
- Evitar situaciones que puedan resultar en lesiones, como practicar deportes.
- evitar procedimientos médicos invasivos como biopsias, trabajos dentales e inmunizaciones intramusculares
- antibióticos profilácticos para proteger contra enfermedades o infecciones cuando sea apropiado
- Medidas de prevención de infecciones, como la higiene adecuada de las manos para protegerse contra enfermedades virales comunes (como la influenza) y otros virus respiratorios, así como complicaciones como la neumonía.
- fisioterapia y terapia ocupacional
- ayudas para la movilidad y otros dispositivos de asistencia como andadores o sillas de ruedas.
- otros dispositivos médicos que pueden ayudar con las actividades de la vida diaria, como vestirse y bañarse
- dispositivos médicos u otras intervenciones de seguridad para ayudar a prevenir caídas, como al levantarse de la cama o ducharse
- programas de asistencia según sea necesario a medida que aumenta la discapacidad
- apoyo psicológico y social para los pacientes y sus familias
- apoyo educativo, incluida la educación especial y la educación en el hogar
- el asesoramiento genético para las familias puede ser útil
No se recomiendan los procedimientos invasivos o cirugías para intentar eliminar áreas de crecimiento óseo anormal, ya que el trauma de la cirugía conduce casi invariablemente al desarrollo de una mayor osificación. Si la cirugía es absolutamente necesaria, se debe utilizar la técnica más mínimamente invasiva posible. Los pacientes con FOP también pueden requerir consideraciones especiales de anestesia.
Se han realizado varios ensayos clínicos en los últimos años con el objetivo de desarrollar mejores opciones de tratamiento para las personas con FOP.
Una palabra de Verywell
La FOP es una afección extremadamente rara en la que una mutación genética hace que los tejidos conectivos del cuerpo, incluidos los músculos, los tendones y los ligamentos, sean reemplazados gradualmente por hueso (osificación). No existe cura para la FOP y diagnosticarla puede ser difícil. El tratamiento es principalmente de apoyo y la progresión de la afección suele ser bastante impredecible. Tomar medidas para evitar lesiones y otras situaciones que aumenten la osificación puede ayudar a reducir la cantidad de "brotes" que tiene una persona, pero aún se puede formar hueso nuevo sin una causa clara. La FOP generalmente conduce a una inmovilidad completa, y la mayoría de las personas se postran en cama a los 30 años. Sin embargo, hay ensayos clínicos en curso que esperan encontrar mejores opciones de tratamiento para mejorar la calidad de vida y los resultados de los pacientes con la afección.
¿Qué sucede cuando el hueso crece dentro del tejido?- Compartir
- Dar la vuelta
- Texto