
Contenido
- Causas
- Los síntomas
- Exámenes y pruebas
- Tratamiento
- Grupos de apoyo
- Perspectiva (pronóstico)
- Posibles complicaciones
- Cuándo contactar a un profesional médico
- Prevención
- Nombres alternativos
- Imágenes
- Referencias
- Fecha de revisión 26/10/2017
La homocistinuria es un trastorno hereditario que afecta el metabolismo del aminoácido metionina. Los aminoácidos son los bloques de construcción de la vida.
Causas
La homocistinuria se hereda en familias como un rasgo autosómico recesivo. Esto significa que el niño debe heredar una copia no funcional del gen de cada padre para que se vea seriamente afectado.
La homocistinuria tiene varias características en común con el síndrome de Marfan, incluyendo cambios en el esqueleto y los ojos.
Los síntomas
Los bebés recién nacidos parecen sanos. Los síntomas tempranos, si están presentes, no son obvios.
Los síntomas pueden aparecer como un retraso leve en el desarrollo o la incapacidad de prosperar. El aumento de los problemas visuales puede conducir al diagnóstico de esta condición.
Otros síntomas incluyen:
- Deformidades del tórax (pectus carinatum, pectus excavatum)
- Enjuague por las mejillas
- Arcos altos de los pies.
- Discapacidad intelectual
- Tocar rodillas
- Extremidades largas
- Desordenes mentales
- Miopía
- Dedos de araña (aracnodactilia)
- Estructura alta y delgada
Exámenes y pruebas
El proveedor de atención médica puede notar que el niño es alto y delgado.
Otros signos incluyen:
- Columna curva (escoliosis)
- Deformidad del pecho.
- Lente dislocada del ojo
Si hay visión deficiente o doble, un oculista (oftalmólogo) realizará un examen de la vista con dilatación para buscar dislocación de la lente o miopía.
Puede haber una historia de coágulos de sangre. Discapacidad intelectual o enfermedad mental también es posible.
Las pruebas que pueden solicitarse incluyen cualquiera de los siguientes:
- Prueba de aminoácidos de la sangre y la orina
- Prueba genética
- Biopsia hepática y ensayo enzimático.
- Radiografía esquelética
- Biopsia de piel con cultivo de fibroblastos.
- Examen oftalmológico estandar
Tratamiento
No existe cura para la homocistinuria. Alrededor de la mitad de las personas con la enfermedad responden a la vitamina B6 (también conocida como piridoxina).
Aquellos que respondan deberán tomar los suplementos de vitamina B6, B9 (folato) y B12 por el resto de sus vidas. Aquellos que no respondan deberán comer una dieta baja en metionina. La mayoría tendrá que ser tratada con trimetilglicina (un medicamento también conocido como betaína).
Ni la dieta ni los medicamentos con bajo contenido de metionina mejorarán la discapacidad intelectual existente. La medicina y la dieta deben ser supervisadas de cerca por un médico con experiencia en el tratamiento de la homocistinuria.
Grupos de apoyo
Estos recursos pueden proporcionar más información sobre la homocistinuria:
- HCU Network America - hcunetworkamerica.org
- NIH / NLM Genetics Home Reference - ghr.nlm.nih.gov/condition/homocystinuria
Perspectiva (pronóstico)
Aunque no existe cura para la homocistinuria, la terapia con vitamina B puede ayudar a aproximadamente la mitad de las personas afectadas por la enfermedad.
Si el diagnóstico se hace en la niñez, comenzar rápidamente una dieta baja en metionina puede prevenir alguna discapacidad intelectual y otras complicaciones de la enfermedad. Por esta razón, algunos estados evalúan la homocistinuria en todos los recién nacidos.
Las personas cuyos niveles de homocisteína en sangre continúan aumentando tienen un mayor riesgo de coágulos de sangre. Los coágulos pueden causar problemas médicos graves y acortar la vida útil.
Posibles complicaciones
Las complicaciones más graves son el resultado de coágulos de sangre. Estos episodios pueden ser potencialmente mortales.
Las lentes dislocadas de los ojos pueden dañar seriamente la visión. La cirugía de reemplazo de lentes puede ser necesaria.
La discapacidad intelectual es una consecuencia grave de la enfermedad. Pero, se puede reducir si se diagnostica temprano.
Cuándo contactar a un profesional médico
Llame a su proveedor si usted o un miembro de su familia muestran síntomas de este trastorno, especialmente si tiene antecedentes familiares de homocistinuria.
Prevención
Se recomienda la asesoría genética para las personas con antecedentes familiares de homocistinuria que desean tener hijos. Se dispone de diagnóstico prenatal de homocistinuria. Esto implica cultivar células amnióticas o vellosidades coriónicas para detectar la cistationina sintasa (la enzima que falta en la homocistinuria).
Si hay defectos genéticos conocidos en los padres o la familia, se pueden usar muestras de muestras de vellosidades coriónicas o amniocentesis para detectar estos defectos.
Nombres alternativos
Deficiencia de cistationina beta-sintasa; Deficiencia de CBS; HCY
Imágenes
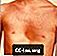
Pectus excavatum
Referencias
Kliegman RM, Stanton BF, St. Geme JW, Schor NF. Defectos en el metabolismo de los aminoácidos. En: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, eds. Nelson Textbook of Pediatrics. 20 ed. Filadelfia, PA: Elsevier; 2016: cap 85.
Schiff M, Blom H. Homocistinuria e hiperhomocisteinemia. En: Goldman L, Schafer AI, eds. Medicina Goldman-Cecil. 25 ed. Filadelfia, PA: Elsevier Saunders; 2016: cap 209.
Fecha de revisión 26/10/2017
Actualizado por: Anna C. Edens Hurst, MD, MS, Profesora Asistente de Genética Médica, Universidad de Alabama en Birmingham, Birmingham, AL. Revisión provista por VeriMed Healthcare Network. También revisado por David Zieve, MD, MHA, Director Médico, Brenda Conaway, Directora Editorial, y el A.D.A.M. Equipo editorial.