
Contenido
- Causas
- Los síntomas
- Exámenes y pruebas
- Tratamiento
- Grupos de apoyo
- Perspectiva (pronóstico)
- Cuándo contactar a un profesional médico
- Prevención
- Nombres alternativos
- Imágenes
- Referencias
- Fecha de revisión 15/10/2018
La enfermedad de Niemann-Pick (NPD, por sus siglas en inglés) es un grupo de enfermedades transmitidas por familias (heredadas) en las que se acumulan sustancias grasas llamadas lípidos en las células del bazo, el hígado y el cerebro.
Hay tres formas comunes de la enfermedad:
- Escribe un
- Tipo B
- Tipo C
Cada tipo involucra diferentes órganos. Puede o no involucrar al sistema nervioso y la respiración. Cada uno puede causar diferentes síntomas y puede ocurrir en diferentes momentos a lo largo de la vida.
Causas
Los tipos A y B de NPD ocurren cuando las células en el cuerpo no tienen una enzima llamada esfingomielinasa ácida (ASM). Esta sustancia ayuda a descomponer (metabolizar) una sustancia grasa llamada esfingomielina, que se encuentra en todas las células del cuerpo.
Si falta ASM o no funciona correctamente, la esfingomielina se acumula dentro de las células. Esto mata las células y dificulta que los órganos funcionen correctamente.
Tipo A se produce en todas las razas y etnias. Es más común en la población judía Ashkenazi (Europa del Este).
El tipo C ocurre cuando el cuerpo no puede descomponer adecuadamente el colesterol y otras grasas (lípidos). Esto conduce a demasiado colesterol en el hígado y el bazo y demasiados lípidos en el cerebro. El tipo C es el más común entre los puertorriqueños de ascendencia española.
El tipo C1 es una variante del tipo C. Se trata de un defecto que interfiere con la forma en que el colesterol se mueve entre las células del cerebro. Este tipo solo se ha visto en personas canadienses francesas en el condado de Yarmouth, Nueva Escocia.
Los síntomas
Los síntomas varían. Otras condiciones de salud pueden causar síntomas similares. Las primeras etapas de la enfermedad solo pueden causar algunos síntomas. Una persona nunca puede tener todos los síntomas.
El tipo A generalmente comienza en los primeros meses de vida. Los síntomas pueden incluir:
- Inflamación abdominal (área abdominal) dentro de los 3 a 6 meses
- Mancha roja cereza en la parte posterior del ojo (en la retina)
- Dificultades de alimentacion
- Pérdida de habilidades motoras tempranas (empeora con el tiempo)
Los síntomas de tipo B suelen ser más leves. Ocurren en la infancia tardía o en la adolescencia. La hinchazón abdominal puede ocurrir en niños pequeños. Casi no hay compromiso del cerebro y del sistema nervioso, como la pérdida de habilidades motoras. Algunos niños pueden tener infecciones respiratorias repetidas.
Los tipos C y C1 suelen afectar a los niños en edad escolar. Sin embargo, puede ocurrir en cualquier momento entre la primera infancia y la edad adulta. Los síntomas pueden incluir:
- Dificultad para mover las extremidades que pueden conducir a una marcha inestable, torpeza, problemas para caminar
- Bazo agrandado
- Hígado agrandado
- Ictericia al nacimiento (o poco después)
- Dificultades de aprendizaje y deterioro intelectual.
- Convulsiones
- Lenguaje confuso e irregular.
- Pérdida repentina del tono muscular que puede provocar caídas.
- Temblores
- Problemas para mover los ojos hacia arriba y hacia abajo
Exámenes y pruebas
Se puede hacer una prueba de sangre o de médula ósea para diagnosticar los tipos A y B. La prueba puede determinar quién tiene la enfermedad, pero no muestra si usted es portador. Se pueden hacer pruebas de ADN para diagnosticar los portadores de los tipos A y B.
Generalmente, se realiza una biopsia de la piel para diagnosticar los tipos C y D. El proveedor de atención médica observa cómo las células de la piel crecen, se mueven y almacenan el colesterol. También se pueden hacer pruebas de ADN para buscar los 2 genes que causan este tipo de enfermedad.
Otras pruebas pueden incluir:
- Aspiración de médula ósea
- Biopsia de hígado (generalmente no es necesaria)
- Examen ocular con lámpara de hendidura
- Pruebas para comprobar el nivel de ASM
Tratamiento
En este momento, no existe un tratamiento eficaz para el tipo A.
Los trasplantes de médula ósea se pueden probar para el tipo B. Los investigadores continúan estudiando posibles tratamientos, incluidos el reemplazo de enzimas y la terapia génica.
Un nuevo medicamento llamado miglustat está disponible para los síntomas del sistema nervioso del tipo C.
El colesterol alto se puede controlar con una dieta o medicamentos saludables y bajos en colesterol. Sin embargo, las investigaciones no muestran que estos métodos eviten que la enfermedad empeore o cambien la forma en que las células descomponen el colesterol. Hay medicamentos disponibles para controlar o aliviar muchos síntomas, como la pérdida repentina del tono muscular y las convulsiones.
Grupos de apoyo
Estas organizaciones pueden brindar apoyo y más información sobre la enfermedad de Niemann-Pick:
- Instituto Nacional de Trastornos Neurológicos y Accidentes Cerebrovasculares: www.ninds.nih.gov/Disorders/All-Disorders/Niemann-Pick-Disease-Information-Page
- Fundación Nacional de Enfermedades de Niemann-Pick - nnpdf.org
- Organización Nacional para los Trastornos Raros - rarediseases.org/rare-diseases/niemann-pick-disease-type-c
Perspectiva (pronóstico)
NPD tipo A es una enfermedad grave. Suele llevar a la muerte a los 2 o 3 años.
Aquellos con el tipo B pueden vivir en la infancia tardía o en la edad adulta.
Un niño que muestra signos de tipo C antes de la edad de 1 año no puede vivir hasta la edad escolar. Aquellos que muestran síntomas después de ingresar a la escuela pueden vivir hasta la adolescencia media o tardía. Algunos pueden vivir hasta los 20 años.
Cuándo contactar a un profesional médico
Haga una cita con su proveedor si tiene antecedentes familiares de la enfermedad de Niemann-Pick y planea tener hijos. Se recomienda asesoramiento genético y screening.
Llame a su proveedor si su hijo tiene síntomas de esta enfermedad, que incluyen:
- Problemas de desarrollo
- Problemas de alimentacion
- Poco aumento de peso
Prevención
Todos los tipos de Niemann-Pick son autosómicos recesivos. Esto significa que ambos padres son portadores. Cada padre tiene 1 copia del gen anormal sin tener ningún signo de la enfermedad en sí.
Cuando ambos padres son portadores, hay un 25% de probabilidad de que su hijo tenga la enfermedad y un 50% de probabilidad de que sea un portador.
Las pruebas de detección de portadores solo son posibles si se identifica el defecto genético. Los defectos involucrados en los tipos A y B han sido bien estudiados. Pruebas de ADN para estas formas de Niemann-Pick están disponibles.
Se han identificado defectos genéticos en el ADN de muchas personas con el tipo C. Es posible que se diagnostiquen las personas que portan el gen anormal.
Algunos centros ofrecen pruebas para diagnosticar a un bebé que todavía está en el útero.
Nombres alternativos
NPD; Deficiencia de esfingomielinasa; Trastorno de almacenamiento de lípidos - enfermedad de Niemann-Pick; Enfermedad de almacenamiento lisosomal - Niemann-Pick
Imágenes
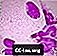
Células espumosas de Niemann-Pick
Referencias
Kliegman RM, Stanton BF, St. Geme JW, Schor NF. Defectos en el metabolismo de los lípidos. En: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, eds. Nelson Textbook of Pediatrics. 20 ed. Filadelfia, PA: Elsevier; 2016: cap 86.
Patterson MC, Hendriksz CJ; Grupo de Trabajo de Directrices NP-C, et al. Recomendaciones para el diagnóstico y tratamiento de la enfermedad de Niemann-Pick tipo C: una actualización. Mol Genet Metab. 2012; 106 (3): 330-344. PMID: 22572546 www.ncbi.nlm.nih.gov/pubmed/22572546.
Turnpenny PD, Ellard S. Errores innatos del metabolismo. En: Turnpenny PD, Ellard S, eds. Elementos de Emery de la genética médica. 15 ed. Filadelfia, PA: Elsevier; 2017: cap 18.
Fecha de revisión 15/10/2018
Actualizado por: Anna C. Edens Hurst, MD, MS, Profesora Asistente de Genética Médica, Universidad de Alabama en Birmingham, Birmingham, AL. Revisión provista por VeriMed Healthcare Network. También revisado por David Zieve, MD, MHA, Director Médico, Brenda Conaway, Directora Editorial, y el A.D.A.M. Equipo editorial.